── Attaching core tidyverse packages ──────────────────────── tidyverse 2.0.0 ──
✔ dplyr 1.1.4.9000 ✔ readr 2.1.5
✔ forcats 1.0.0 ✔ stringr 1.5.1
✔ ggplot2 3.5.1 ✔ tibble 3.2.1
✔ lubridate 1.9.3 ✔ tidyr 1.3.1
✔ purrr 1.0.2
── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()
ℹ Use the conflicted package (<http://conflicted.r-lib.org/>) to force all conflicts to become errors
# Set the seed for reproducibilityset.seed(20240425)
31.1 Exponential Distribution
The exponential distribution is commonly used as a waiting time distribution, i.e. the time until something happens.
31.1.1 Probability density function
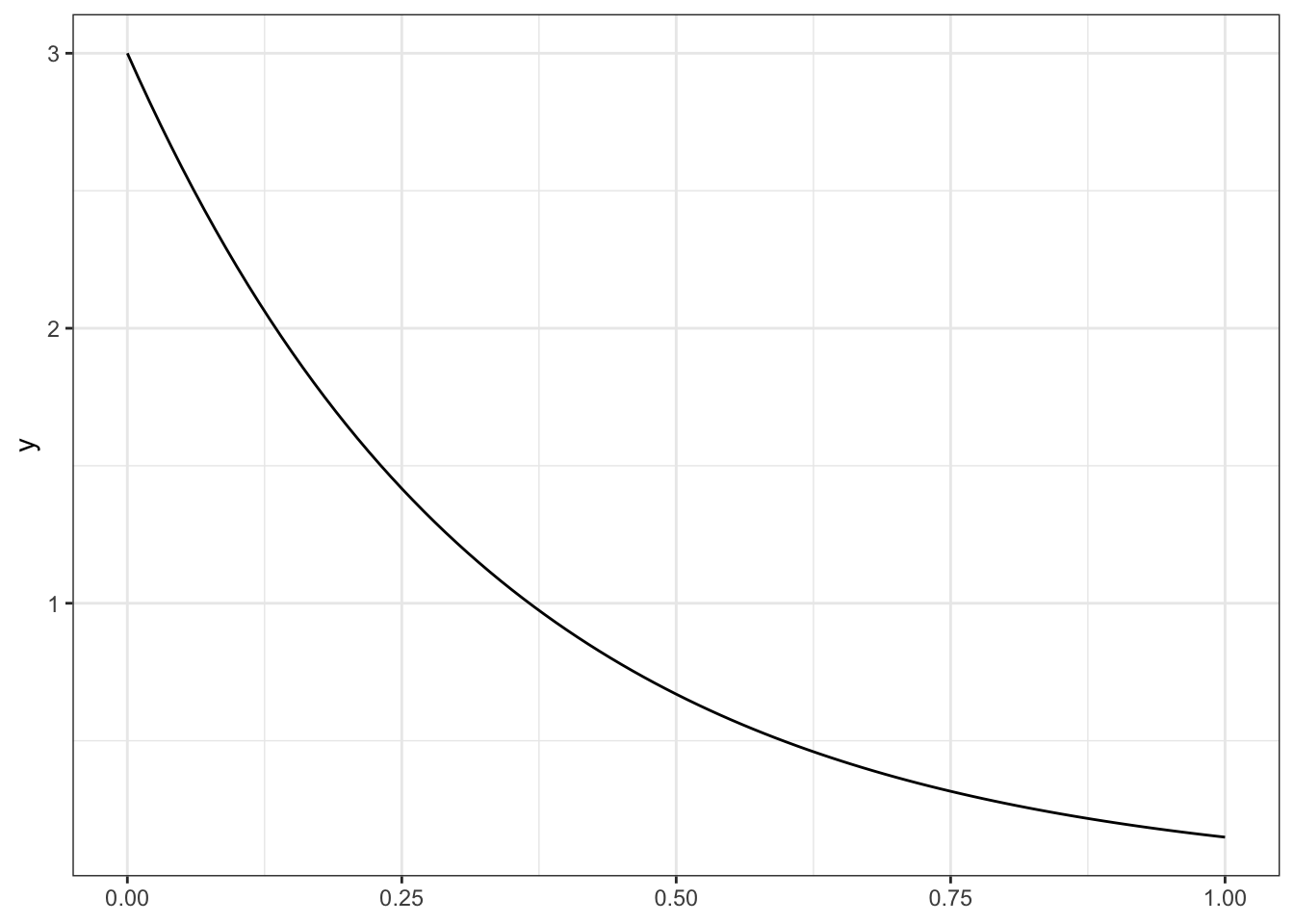
A random variable \(X\) has an exponential distribution if its density function is \[
f(x) = \lambda e^{-\lambda x} \quad \mbox{for }x>0
\] for rate \(\lambda>0\).
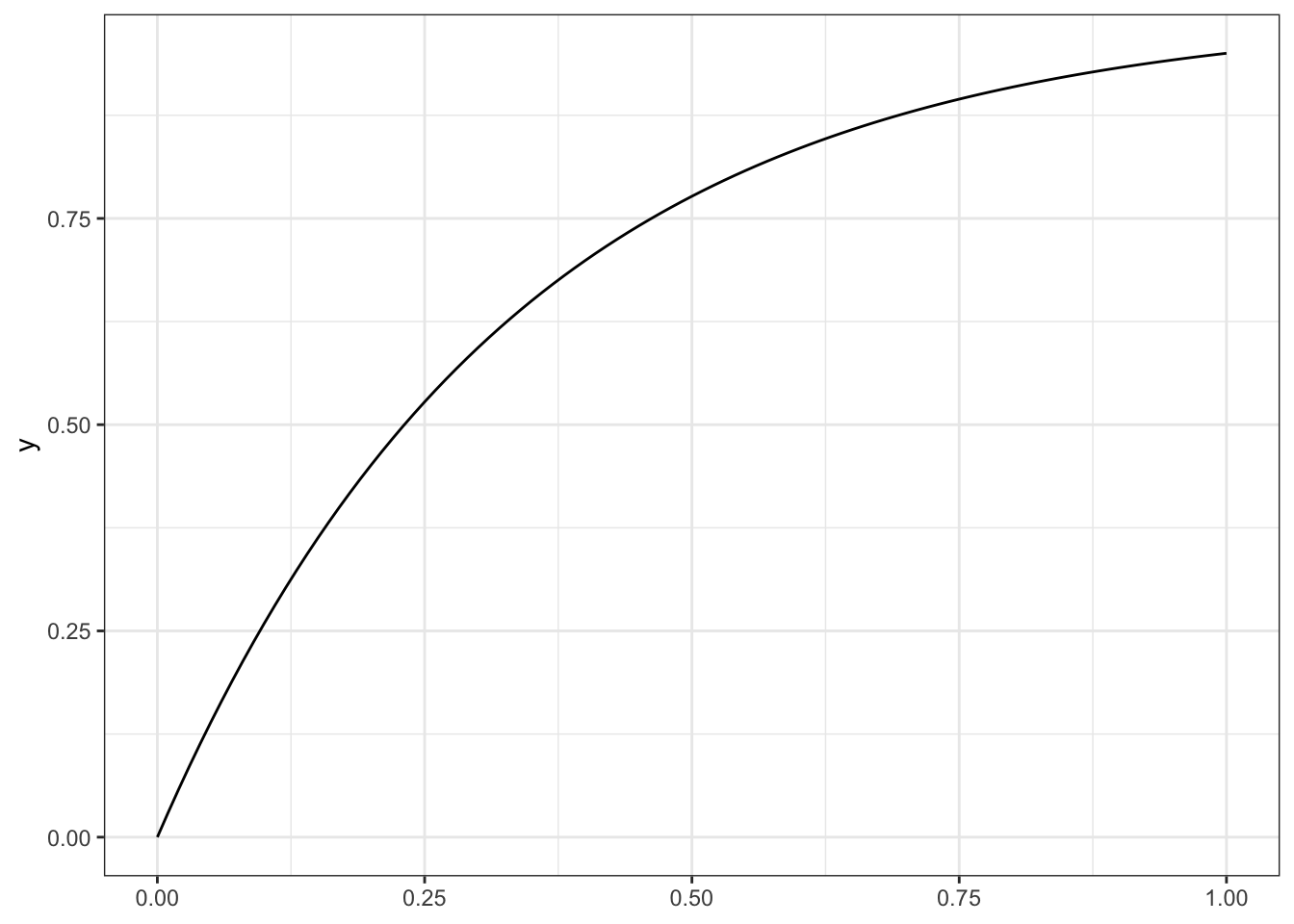
The probability of being greater than some value has a simple form for the exponential distribution. \[
P(X > x) = 1-P(X\le x) = e^{-\lambda x}.
\]
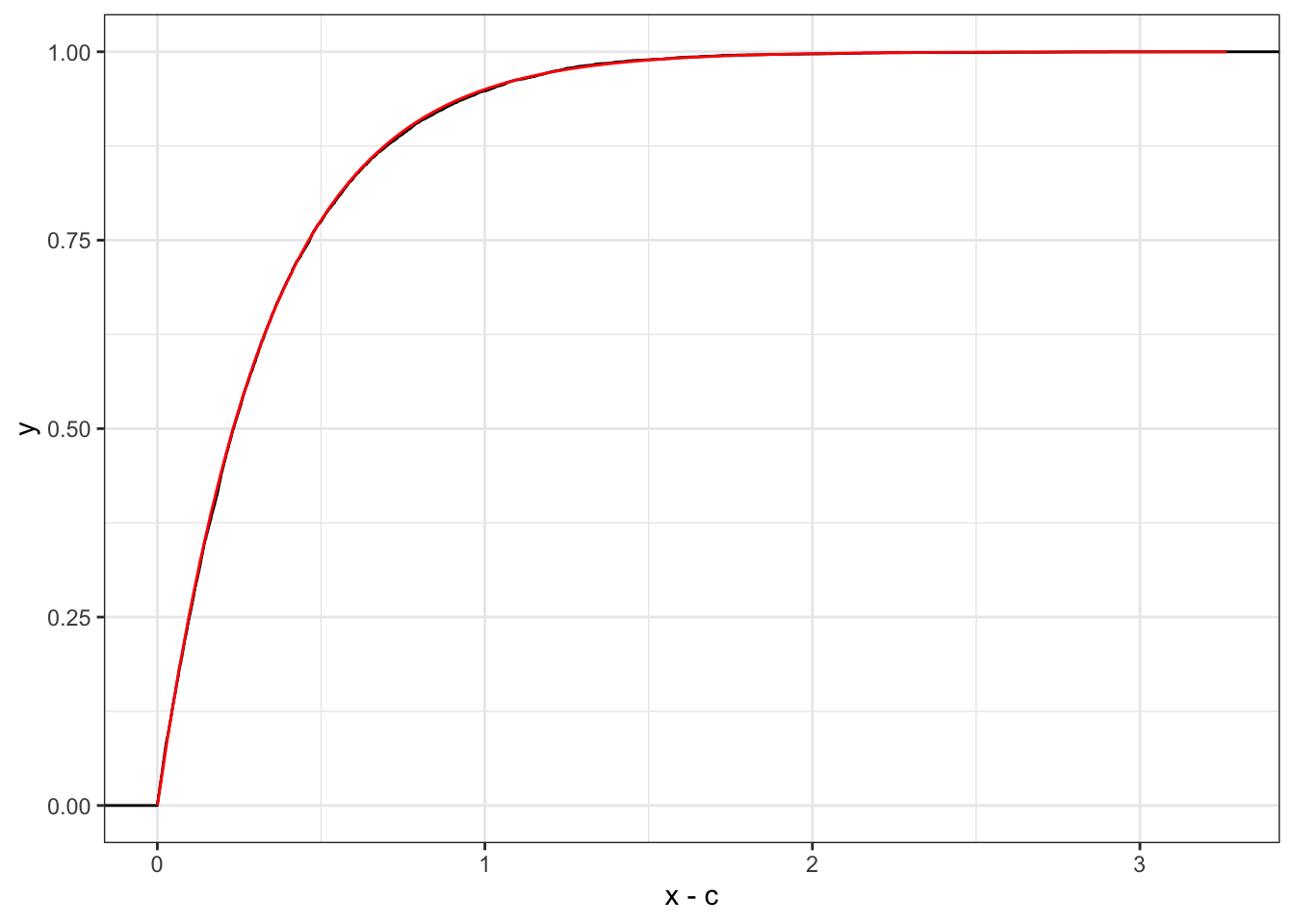
The exponential distribution has a property called the memoryless property that states \[
P(X > x+c | X > c) = P(X > x) = e^{-\lambda x}
\]
For example,
# Exponential conditionalc <-1# Check probability 1-pexp(c, rate = r) # if this probability is too small, the simulation will take a while
[1] 0.04978707
# Simulate exponential random variables that are greater than cx <-rexp(1e4, rate = r)for (i inseq_along(x)) {while (x[i] < c) x[i] <-rexp(1, rate = r)}
# Plot density for conditionalggplot(data.frame(x = x), aes(x - c) # performs the shift) +stat_ecdf() +stat_function(fun = pexp, args =list(rate = r), color ="red")
31.1.4 Minimum exponential
Let \[
X_i \stackrel{ind}{\sim} Exp(\lambda_i)
\] what is the distribution for \(T = \min\{X_1,\ldots,X_n\}\)?
It turns out that \[
T \sim Exp\left(\sum_{i=1}^n \lambda_i\right).
\]
If you are only told that \(T=t\) which \(X_i=t\)?
That is, which exponential was the minimum one?
We won’t know, but the probability is proportional to the rate. So, we know \[
P(X_i = t) \propto \lambda_i = \frac{\lambda_i}{\sum_{j=1}^n \lambda_j}.
\]
31.2 Continuous time Markov process
These types of models are used in a wide variety of fields. All of these a stochastic models that utilize a set of transitions between states that involve simulating exponential random variables for the time of the next transition and which transition occurs.
31.2.1 Michaelis-Menton kinetics
Here we will motivate the module using a chemistry experiment called Michaelis-Menton kinetics. This experiment creates a product “P” from a substrate “S” in a reaction that is catalyzed by an enzyme “E”. In order to convert the substrate to the product, the enzyme must bind to the substrate to create the enzyme-substrate complex “ES”.
This system can be represented by this system of reactions \[
E + S \leftrightarrows ES \to E + P.
\] This system of reactions actually has 3 reactions: \[ \begin{array}{rll}
I:& E + S &\stackrel{\lambda_1}{\to} ES \\
II:& ES &\stackrel{\lambda_2}{\to} E + S \\
III:& ES &\stackrel{\lambda_3}{\to} E + P \\
\end{array} \] The \(\lambda\) indicate the rate of the reaction.
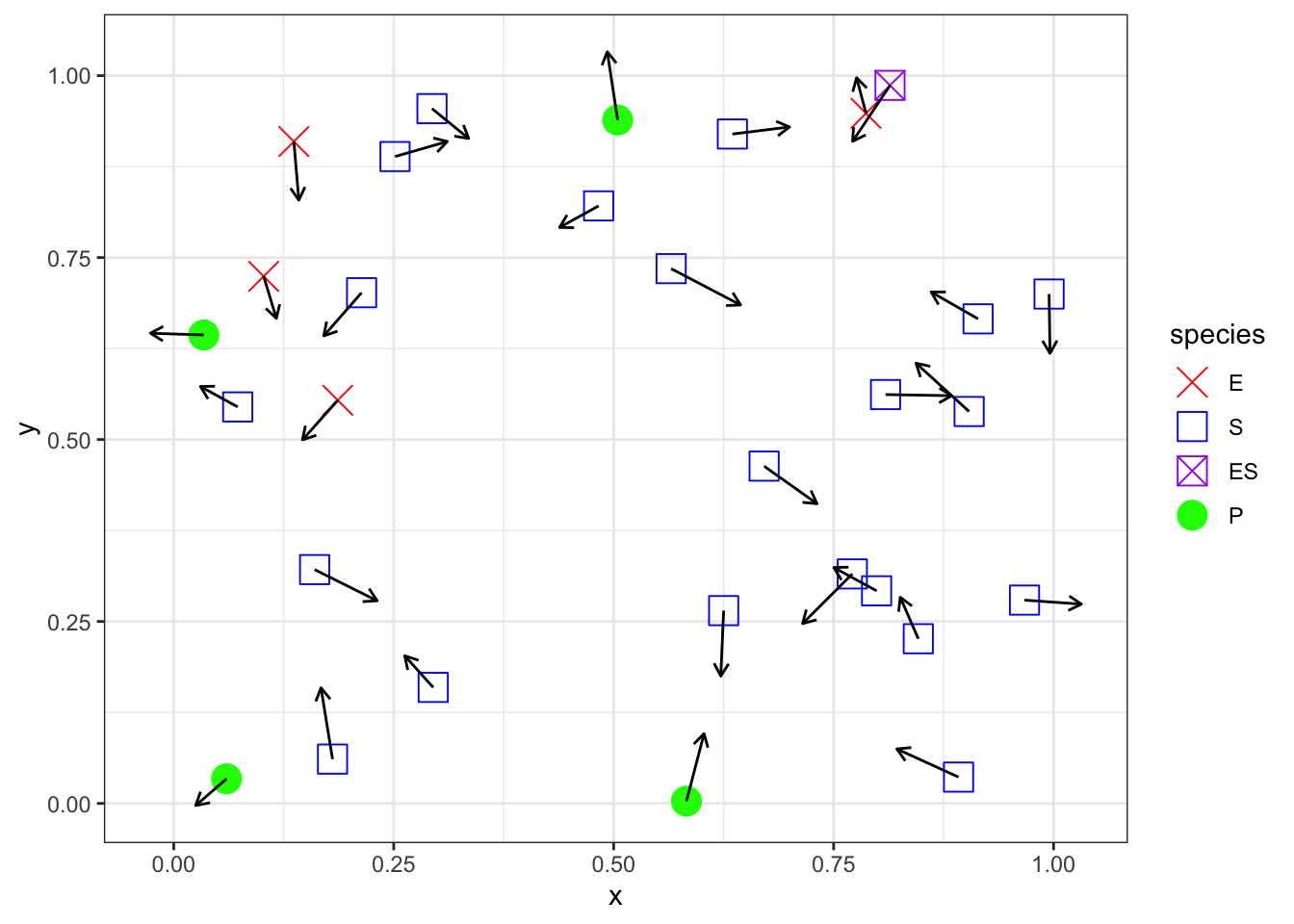
In this system, reaction I requires that an enzyme molecule come into contact with a substrate molecule.
If these molecules are in a volume, then the system might look like this
set.seed(20230309)n <-30d <-data.frame(x =runif(n),y =runif(n),species =sample(c("E","S","ES","P"), size = n, replace =TRUE, prob =c(0.2,0.6,0.1,0.1)),speed =runif(n,0.05, 0.1),direction =runif(n, max =2*pi)) %>%mutate(xend = x + speed*cos(direction),yend = y + speed*sin(direction),species =factor(species, levels =c("E","S","ES","P")) )ggplot(d, aes(x = x, y = y, xend = xend, yend=yend,color = species, shape = species)) +geom_point(size =5) +geom_segment(arrow =arrow(length =unit(0.2, "cm")), color ="black") +scale_color_manual(values =c("E"="red","S"="blue","ES"="purple","P"="green" ) ) +scale_shape_manual(values =c("E"=4,"S"=0,"ES"=7,"P"=19 ) )
These reactions can be coded by indicting two matrices: Pre and Post. These matrices indicate the reactants (Pre) and the products (Post).
Pre <-rbind(# E S ES Pc(1, 1, 0, 0),c(0, 0, 1, 0),c(0, 0, 1, 0))rownames(Pre) <-c("I","II","III")colnames(Pre) <-c("E","S","ES","P")Pre
E S ES P
I 1 1 0 0
II 0 0 1 0
III 0 0 1 0
Post <-rbind(# E S ES Pc(0, 0, 1, 0),c(1, 1, 0, 0),c(1, 0, 0, 1))rownames(Post) <-c("I","II","III")colnames(Post) <-c("E","S","ES","P")Post
E S ES P
I 0 0 1 0
II 1 1 0 0
III 1 0 0 1
The reaction matrix is constructed by subtracting these and the stoichiometry matrix by transposing the reaction matrix.
A <- Post - Pre # reactionS <-t(A) # stoichiometryA
E S ES P
I -1 -1 1 0
II 1 1 -1 0
III 1 0 -1 1
S
I II III
E -1 1 1
S -1 1 0
ES 1 -1 -1
P 0 0 1
We can update the state of the system by either the stoichiometry or reaction matrix.
x0 <-c(5, 100, 1, 0)x0 + S[,1]
E S ES P
4 99 2 0
x0 + S[,2]
E S ES P
6 101 0 0
x0 + S[,3]
E S ES P
6 100 0 1
To determine which reaction occurs we need to calculate the propensity of the reaction. The propensity is the product of two components: the reaction rate and the number of combinations of the Pre molecules.
For this model we have the following propensities: \[ \begin{array}{rll}
I:& \lambda_1 E \cdot S \\
II:& \lambda_2 ES \\
III:& \lambda_3 ES \\
\end{array} \]
31.2.1.1 Monte Carlo Simulation
To simulate this continuous time Markov process, we iterate through these steps
Simulate a time increment which has an exponential distribution with rate equal to the sum of the propensities.
Simulate which reaction occurred which has a discrete distribution with probability proportional to the propensity.
Update time according to the time increment.
Update the state according to the reaction/stoichiometry matrix
31.2.1.2 R implementation
michaelis_menton_reaction_matrix <-rbind(# E S ES Pc(-1, -1, 1, 0),c( 1, 1, -1, 0),c( 1, 0, -1, 1))#' Function to simulate a single Michaelis-Menton transition#' #' This function will simulate a single predator-prety transition based on the #' current `state` of the system and the reaction `rates`simulate_michaelis_menton_transition <-function(state, rate, initial_time) { propensity <-c( rate[1] * state[1] * state[2], rate[2] * state[3] , rate[3] * state[3] ) time_increment <-rexp(1, rate =sum(propensity)) reaction <-sample(1:3, 1, prob = propensity) state <- state + michaelis_menton_reaction_matrix[reaction,]return(list(time = initial_time + time_increment,state = state ))}
simulate_michaelis_menton_system <-function(initial_state, rate, max_rxns =1e3) {# Create storage structures for state and time n_possible_rxns <- max_rxns +1 state <-matrix(initial_state, nrow =4, ncol = n_possible_rxns) n_rxns <-1 time <-rep(0, n_possible_rxns) # Simulate outbreakwhile(any(state[2:3,n_rxns] >0) & n_rxns <= max_rxns) { tmp <-simulate_michaelis_menton_transition(state[,n_rxns], rate, time[n_rxns]) n_rxns <- n_rxns +1 time[n_rxns] <- tmp$time state[,n_rxns] <- tmp$state } d <-data.frame(E = state[1, ],S = state[2, ],ES = state[3, ],P = state[4, ],time = time ) %>%pivot_longer(-time, names_to ="state", values_to ="count") %>%mutate(state =factor(state, levels =c("E","S","ES","P")) )}
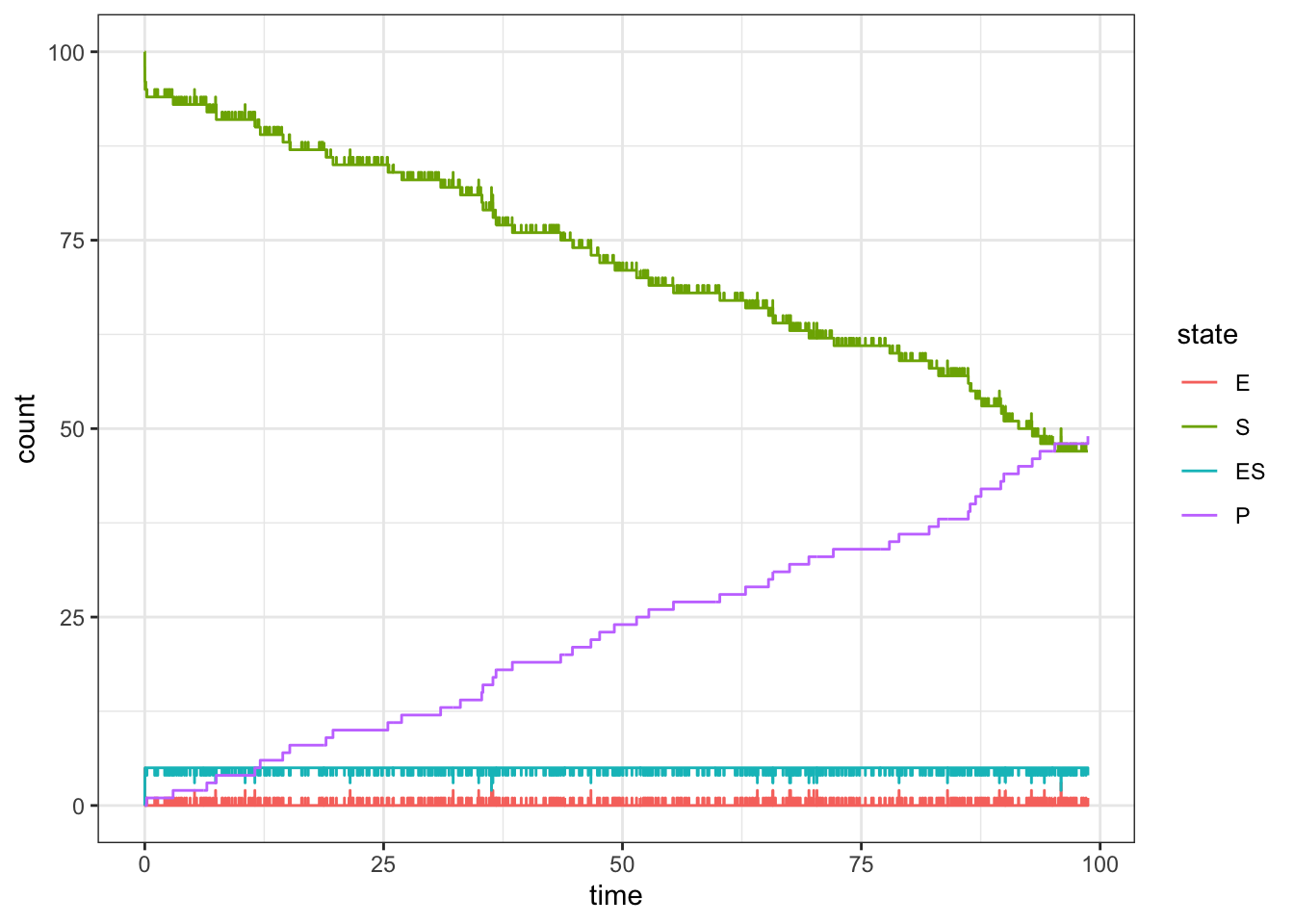
set.seed(1)d <-simulate_michaelis_menton_system(c(5, 100, 0, 0), rate =c(1,1,.1), max_rxns =1e3)ggplot(d, aes(x = time, y = count, color = state)) +geom_step()
This model is a continuous time Markov process because
Time can be incremented by any amount (rather than a discrete amount).
The state of the system is updated based on the current state (as opposed to past history).
31.2.2 SIR Model
The SIR model has three states:
[S]usceptible
[I]nfectious
[R]ecovered
There are two reactions that can occur \[ \begin{array}{r\quad{}ll}
I:&S + I &\stackrel{\lambda}{\to} 2I \\
II:&I &\stackrel{\rho}{\to} R
\end{array} \]
The propensity of these reactions are \[ \begin{array}{r\quad{}ll}
I:& \lambda \, S\cdot I / N \\
II:& \rho \, I
\end{array} \]
The system is updated by the reaction matrix
\[ \begin{array}{r|rrr}
& S & I & R \\
\hline
I & -1 & 1 & 0 \\
II & 0 & -1 & 1\\
\end{array} \]
Due to the two reactions converting a single individual from one state to another, this model has a constant population size.
31.2.2.1 R implementation
sir_reaction_matrix <-rbind(# S I Rc(-1, 1, 0),c( 0, -1, 1))#' Function to simulate a single SIR transition#' #' This function will simulate a single SIR transition based on the current #' `state` of the system and the reaction `rates`simulate_sir_transition <-function(state, rate, initial_time) { propensity <-c( rate[1] * state[1] * state[2] /sum(state), rate[2] * state[2] ) time_increment <-rexp(1, rate =sum(propensity)) reaction <-sample(1:2, 1, prob = propensity)# Update state according to stoichiometry state <- state + sir_reaction_matrix[reaction,]return(list(time = initial_time + time_increment,state = state ))}
simulate_outbreak <-function(initial_state, rate, max_rxns =5000) {# Create storage structures for state and time n_possible_rxns <-min(2*initial_state[1] + initial_state[2]+1, max_rxns +1) state <-matrix(initial_state, nrow =3, ncol = n_possible_rxns) n_rxns <-1 time <-rep(0, n_possible_rxns) # Simulate outbreakwhile(state[2,n_rxns] >0& n_rxns <= max_rxns ) { tmp <-simulate_sir_transition(state[,n_rxns], rate, time[n_rxns]) n_rxns <- n_rxns +1 time[n_rxns] <- tmp$time state[,n_rxns] <- tmp$state }# Clean up data frames n <-which(state[2,] ==0) d <-data.frame(S = state[1, 1:n],I = state[2, 1:n],R = state[3, 1:n],time = time[1:n] ) %>%pivot_longer(-time, names_to ="state", values_to ="count") %>%mutate(state =factor(state, levels =c("S","I","R")) )}
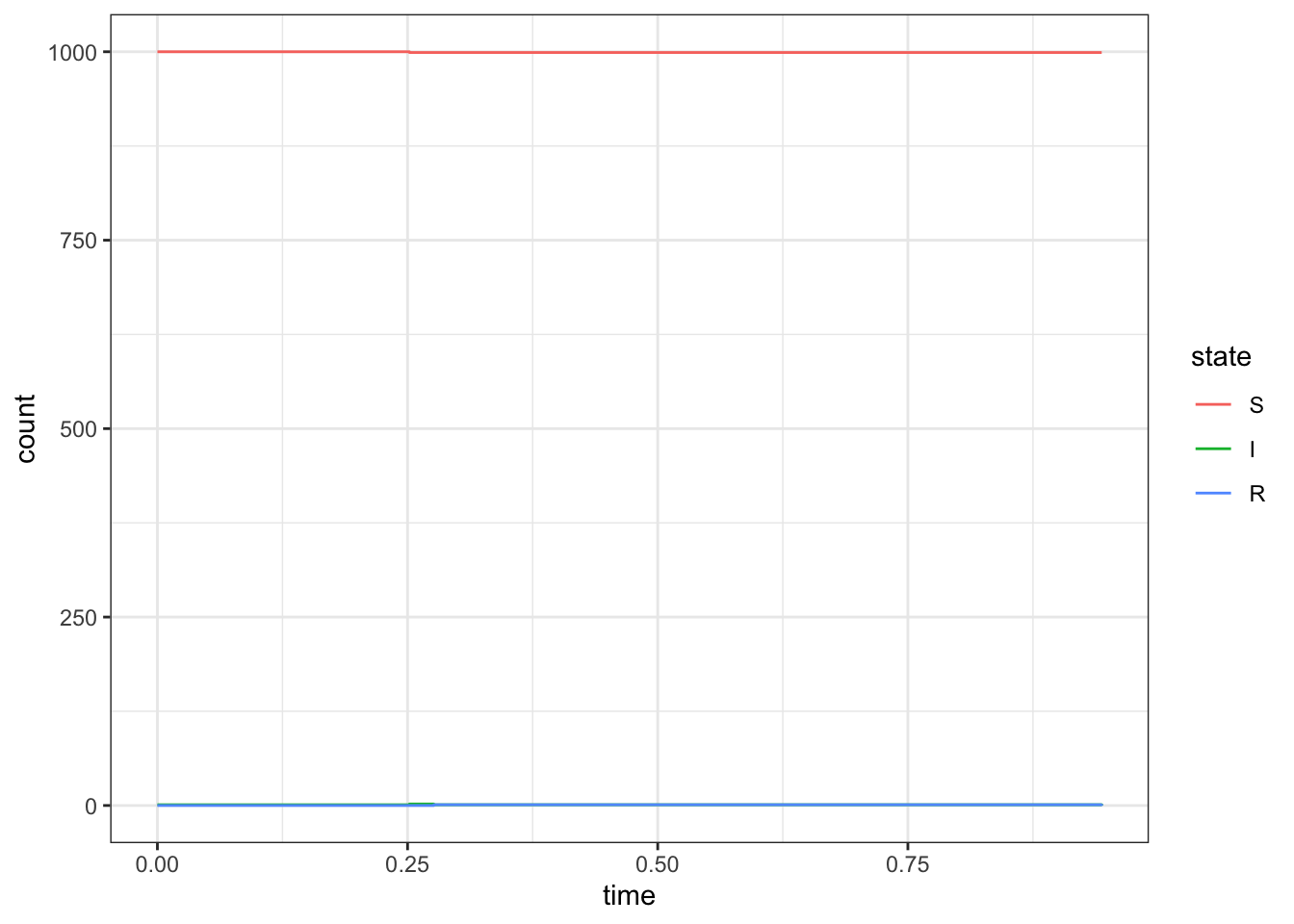
Sometimes the outbreak will die out right away
set.seed(1)d <-simulate_outbreak(c(1000, 1, 0), rate =c(2,1))ggplot(d, aes(x = time, y = count, color = state)) +geom_step()
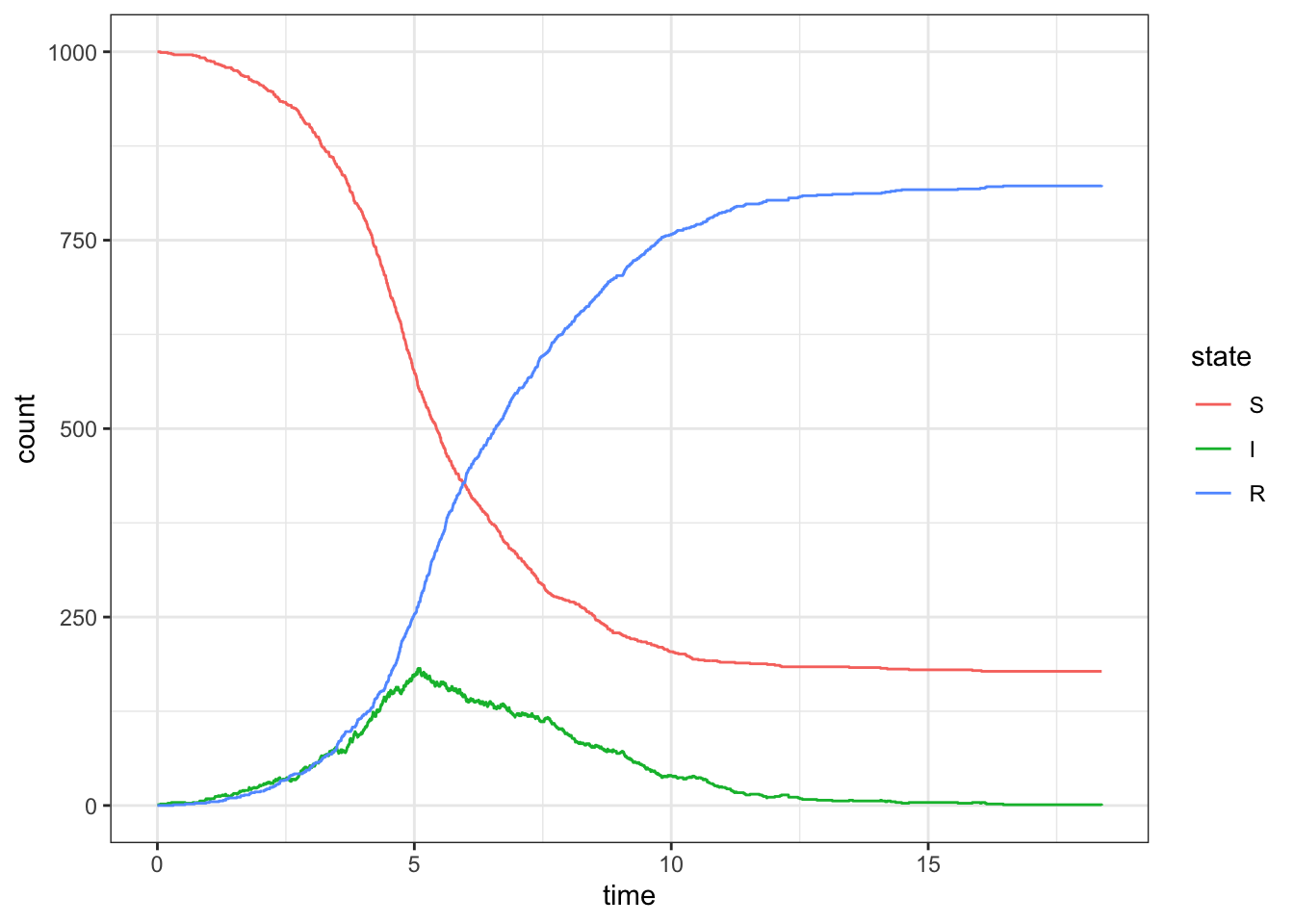
Sometimes the outbreak will last a while
set.seed(4)d <-simulate_outbreak(c(1000, 1, 0), rate =c(2,1))ggplot(d, aes(x = time, y = count, color = state)) +geom_step()
31.2.2.2 Scientific questions
One question we might ask is: what is the probability the outbreak will die out quickly (before time 5) for a given initial state and reaction rates.
To answer this question, we can run a Monte Carlo study
Another question could be, given the outbreak lasts longer than 5, what is the expected number of people who will be infected?
X0 <-c(1000, 1, 0)rate <-c(2,1)infected <-replicate(1e3, { d <-simulate_outbreak(X0, rate)while(max(d$time) <5) d <-simulate_outbreak(X0, rate)return(max(d$count[d$state =="R"]))})t.test(infected)$conf.int # MC estimate of
A simple predator-prey model (stochastic version of the Lotka-Volterra Model) has two states:
[R]abbits
[F]oxes
There are two reactions that can occur \[ \begin{array}{r\quad{}ll}
I:&R &\stackrel{\lambda}{\to} 2R \\
II:&F+R &\stackrel{\rho}{\to} 2F \\
II:&F &\stackrel{\delta}{\to} \emptyset \\
\end{array} \] The first reaction simply
The propensity of these reactions are \[ \begin{array}{r\quad{}ll}
I:& \lambda \, R \\
II:& \rho \, F\cdot R\\
II:& \delta \, F
\end{array} \]
The system is updated by the reaction matrix
\[ \begin{array}{r|rrr}
& R & F \\
\hline
I & 1 & 0 \\
II & -1 & 1 \\
III & 0 & -1
\end{array} \]
This model does not have a constant population size.
31.2.3.1 R implementation
predator_prey_reaction_matrix <-rbind(# R Fc( 1, 0),c(-1, 1),c( 0, -1))#' Function to simulate a single predator-prey transition#' #' This function will simulate a single predator-prety transition based on the #' current `state` of the system and the reaction `rates`simulate_transition <-function(state, rate, initial_time) { propensity <-c( rate[1] * state[1], rate[2] * state[1] * state[2], rate[3] * state[2] ) time_increment <-rexp(1, rate =sum(propensity)) reaction <-sample(1:3, 1, prob = propensity)# Update state according to reaction matrix state <- state + predator_prey_reaction_matrix[reaction,]return(list(time = initial_time + time_increment,state = state ))}
simulate_predator_prey_system <-function(initial_state, rate, max_rxns =1e4) {# Create storage structures for state and time n_possible_rxns <- max_rxns +1 state <-matrix(initial_state, nrow =2, ncol = n_possible_rxns) n_rxns <-1 time <-rep(0, n_possible_rxns) # Simulate outbreakwhile(state[2,n_rxns] >0& n_rxns <= max_rxns ) { tmp <-simulate_transition(state[,n_rxns], rate, time[n_rxns]) n_rxns <- n_rxns +1 time[n_rxns] <- tmp$time state[,n_rxns] <- tmp$state } d <-data.frame(R = state[1, ],F = state[2, ],time = time ) %>%pivot_longer(-time, names_to ="state", values_to ="count") %>%mutate(state =factor(state, levels =c("R","F")) )}
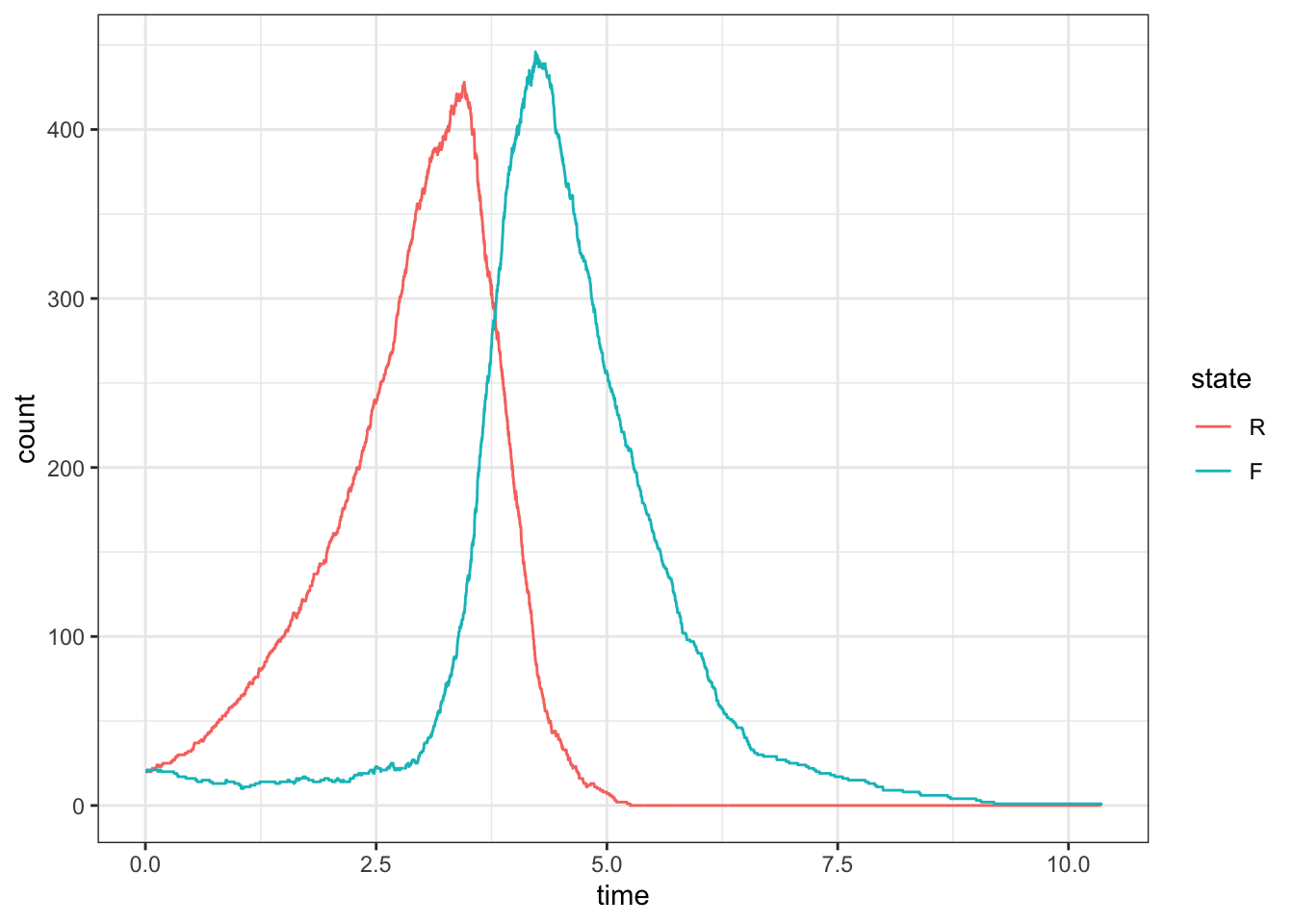
Sometimes the foxes will eat all the rabbits
set.seed(1)d <-simulate_predator_prey_system(c(20,20), rate =c(1,.01,1), max_rxns =1e5)ggplot(d, aes(x = time, y = count, color = state)) +geom_step()